2020IGEM
3Dmol.js
蛋白质三级结构比对概述
蛋白质结构以及分类
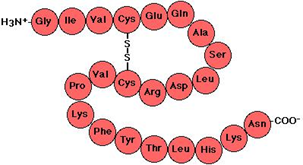
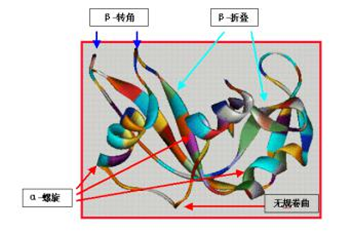
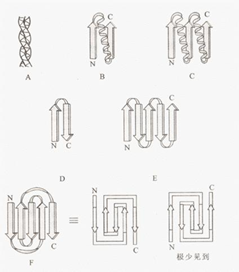
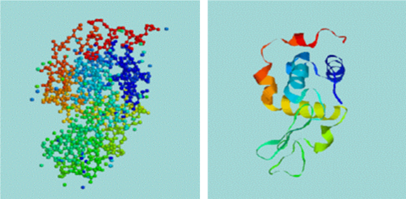
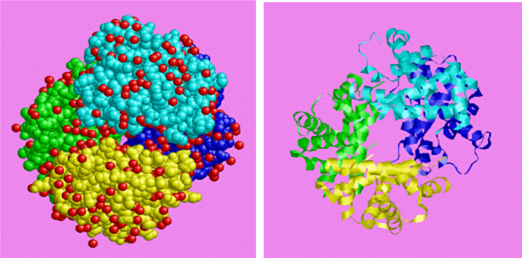
研究蛋白质结构比对的意义
- 一种功能未知但结构已知的蛋白质能与功能和结构都已知的蛋白质进行比对。这种比对可以提供关于该蛋白质功能的线索。
- 结构比对可以用来对蛋白质进行分类。由于蛋白质的结构与其功能紧密相关,所以基于结构的分类方法具有很重要的生物学意义。
- 对于一组功能相似的蛋白质进行多重结构比对可以鉴别出与该功能相关的保守结构片段。
- 可以利用结构比对发现远程同源关系。
数据库
PDB数据库
PDB(Protein Data Bank)是一个包含蛋白质和核酸等生物大分子的数据库。用户可直接查询、调用和观察库中所收录的任何大分子三维结构,它包含的蛋白质和核酸三维结构资料为新药物、生物燃料、纳米材料的设计以及生物学和医学方面的基本发现提供了非常重要的资源。
- 能够查找目的蛋白质的结构数据;
- 可以对蛋白质四个级别的结构进行分析;
- 链接如 GDB、GenBank、SWISS-PROT、PIR 等互联网数据库,通过这些数据库查询蛋白质的其他信息;
- 可下载有关的结构信息以供进一步使用,可通过关键词或 PDB 标识符等进行查询;
- PDB 通过对蛋白质的序列进行分析,用于结构预测和同源性比较。
相似度比较方法
为什么要基于三维结构:
(1)一些相同的氨基酸序列在不同条件下可能折叠成具有不同功能的空间构象,例如天然型朊蛋白与感染型朊蛋白,呈螺旋结构的天然型朊蛋白在动物体内维持正常的生理功能,在特殊情况下,天然型朊蛋白可能会转变为折叠形状的感染型朊蛋白,从而导致某些如老年痴呆等神经性疾病,若使用序列分析的方法,找不出它们之间的差异,只有通过三维结构的比较才能得到它们之间的不同,进而分析功能不同的原因。
(2)有些蛋白质序列的个别氨基酸发生突变,也可能导致其结构与功能发生变化,最典型的案例是镰刀形细胞贫血症,它是由于组成人类血红蛋白链上146 个氨基酸中的第 6 个氨基酸谷氨酸被替换成了缬氨酸,使正常的圆盘状的红细胞,变为镰刀状,导致携带氧的功能减为正常红细胞的一半。此外,蛋白质 erbb4 羧基尾端第 1045 和 1139 两个氨基酸发生突变,使用从头建模的建模工具 I-TASSER 和 QUARK对突变前后的包含 20 个氨基酸的片段进行建模,发现第 1045 和 1139 个氨基酸片段从螺旋变为折叠,改变了它在胃肿瘤组织中的致癌作用。
(3)很多序列相关性很低、甚至序列无关的蛋白质在其空间三维结构上却
有着惊人的相似之处,对蛋白质三维结构的比较方法可以检测到序列同源性相
距甚远的蛋白质之间的结构相似性,这可以帮助生物学者发现长期的进化关
系,进一步理解进化的机制,找到远程的具有同源关系的蛋白质。
(4)通过对蛋白质三维结构的分析,可以确定与某些功能相关的子结构或
模体,依据这些子单元来研究蛋白质结构中的作用机制,从而进行药物的设计,
对疾病进行预防和治疗。
mTM-align
蛋白质三级结构比对原理
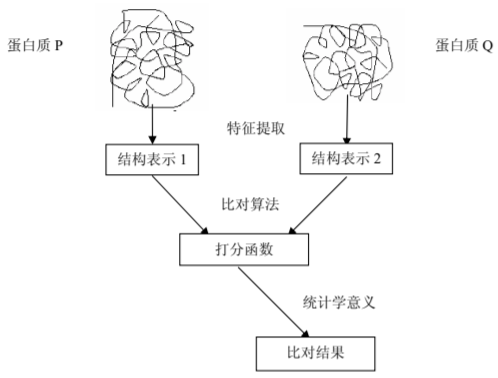
评价指标
多重结构比对算法的评价指标
全局的评价指标
全局的评价指标建立在从多重结构比对中导出的两两结构比对之上。
- 匹配的残基对的数量: 如果在结构旋转后,两个对应上的残基之间的距离小于 4Å (0.4nm),则称他们为结构上匹配的残基对;计算两两比对中匹配的残基对的数量,记作 $L_{ali}$
- RMSD 均方根差 量测重叠蛋白质分子间的距离
- TM-score = $max(\frac{1}{L_N}\displaystyle \sum^{L_T}_{i=1}{\frac{1}{1+(\frac{d_i}{d_0})^2}})$ 值越大,相似度越高
- 比对的准确率(ACC): 该两两比对与参考比对相同的匹配的残基对的数量除以参考比对的匹配的残基对的数量。
- $RT$: running time 运行时间
局部的评价指标
局部的评价指标建立在共同核心区域(common core)之上。
核心区域(common core):1.该列中没有空格;或者2.经过旋转后,该列中的残基两两之间的距离均小于 4 Å。 则称这一列为共同核心区域。
- $L_{core}$:共同核心区域的长度。
- $ccRMSD$:共同核心区域上的平均均方根偏差。
- $ccTM-score$: 共同核心区域上的平均TM-score。
- $RT$: running time 运行时间
结构数据库的快速搜索算法的指标
c(n):
$$c(n)=\frac{1}{T} \sum_{i=1}^{T}\left(\frac{1}{\min \left(N_{i}, q\right)} \sum_{i=1}^{\min \left(N_{i}, q\right)} c_{q}\left(q_{i}, t\right)\right)$$
$c_q(q, t)$: 对于两个结构$q$和$t$ ,STOVCA将从两两结构比对算法给出的比对中导出一个最优的旋转方式,从而给出一系列衡量比对质量的指标。平均准确率p(n)和平均召回率r(n):
是基于 SCOP 数据库中关于折叠类型的定义。
如果在查询结构返回的搜索结果中,一个结构与查询结构处于同一个折叠类型中,那么这个结构被认为是真阳性的。如果查询结构返回的搜索结果中有结构没有在 SCOP 数据库中的注释,则不将该结构纳入计算范畴。
$$\begin{array}{l}
p(n)=\frac{1}{500} \sum_{i=1}^{500} \frac{T P\left(m_{i}\right)}{m_{i}} \
r(n)=\frac{1}{500} \sum_{i=1}^{500} \frac{T P\left(m_{i}\right)}{P_{i}}
\end{array}$$$RT$: running time 运行时间
TM-align 算法
旨在找到使得两个结构 TM-score 得分最高的残基之间的对应关系
步骤: 初始化比对 && 进行启发式迭代
初始化比对
5种方法
- 无空格穿线法(gaplessthreading,记作 IA1),
- 基于二级结构的方法(secondary structure,记作 IA2),
- 基于二级结构优化IA1的方法(记作IA3),
- 局部蛋白质叠加法(localstructure superposition,记作 IA4),
- 基于碎片的无空格穿线法(fragment-based gaplessthreading,记作IA5)。
确定初始比对后,将每种初始比对代入迭代算法,取经过迭代后 TMscore 最高的结果为最终结果。
启发式迭代
首先基于初始比对中残基的对应关系旋转结构,相似性打分矩阵定义如下:
$$s(i, j) = \frac{1}{1+\frac{d_{ij}^2}{d_0^2}}$$
$d_{ij}$表示在经过旋转后,结构 1 中第$i$ 个残基和结构 2 中第 $j$ 个残基间的距离。以 $s(i,j)$为打分矩阵和 0.6 为空位开启罚分(gap penalty)进行动态规,可得到一个新的比对。根据新的比对进行旋转,再根据新的相似性打分矩阵开始实施动态规划,得到更新的结构比对。这个过程一直进行下去,直到比对结果稳定并且返回一个最高的 TM-score 分值。基于 TM-score 相似性打分的合理性,算法收敛速度很快,通常进行 2~3 次迭代就能得到最优比对。
mTM-align 多重结构比对算法流程
给定一组蛋白质结构,mTM-align算法将通过下述三个步骤构建多重结构比对: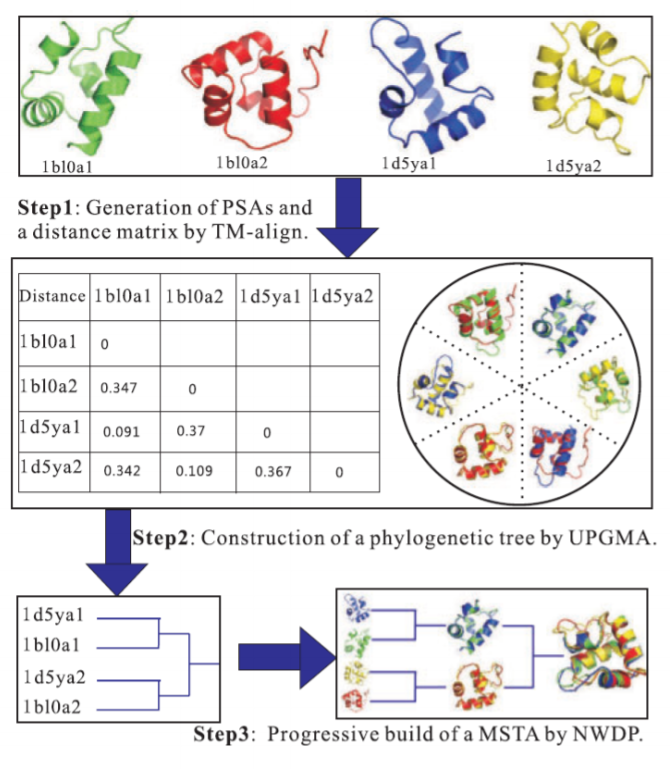
step1:通过 TM-align 进行两两结构比对;
step2:构建进化树:
为了使蛋白质结构可以进行有序的逐步比对,需要通过多结构之间的进化关系来确定逐步比对的顺序。因此需要构建进化树。
step3:构建多重结构比对。
根据进化树从叶子到根的顺序,逐步进行多重结构比对。
如果是两个结构进行比对,则将直接使用第一步中 TM-align 的结果。如果比对在两个已构建好的比对之间进行,或者是在一个已构建好的比对和一个结构之间进行,则将采
用 Needleman-Wunsch的动态规划(Dynamic Programming,DP)算法。
结构数据库的快速搜索算法流程
数据库处理
蛋白质链数据库(PDB chain,PDBC)
蛋白质链数据库是将蛋白质数据库中所有蛋白质结构按链拆分,然后去除长度小于 10 个残基的链,最终得到约 40 万条蛋白质链。
蛋白质结构域数据库(Domain database,DOM)
分为两个部分,一部分是直接获取SCOP数据库中有的结构域信息,另一部分是针对在蛋白质数据库中存在,而 SCOP数据库中没有信息的蛋白质链,使用 PDP程序将其切分为蛋白质结构域。然后去除所有蛋白质结构域中长度小于 10 个残基的结构域,最终得到了大约 50 万个结构域。
mTM-align的效果
举例
HOMSTRAD 数据集中‘YgbB’蛋白质家族,该家族中有三个结构。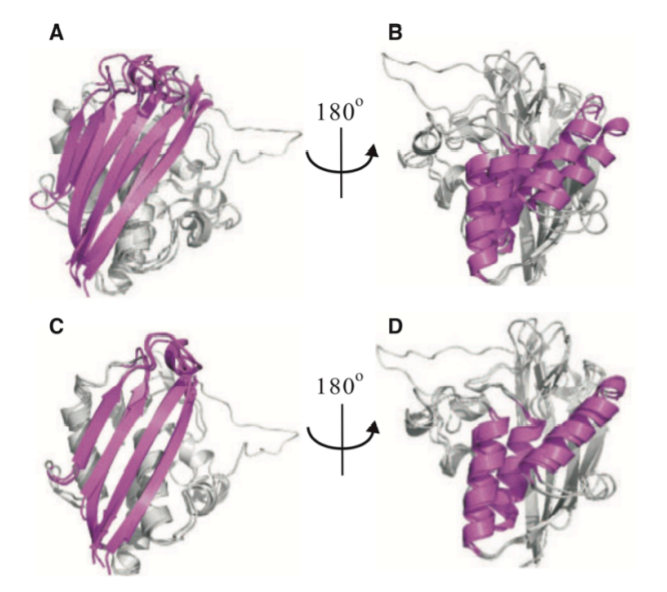
结论
mTM-align可以为更广泛的应用提供更可靠的结构比对。
服务器的输入和输出
服务器的输入
- 结构数据库搜索模块
输入为一个 PDB 格式的蛋白质结构,输入的结构只能包含一条链。
PDB数据库:保存的是每个蛋白质的原子坐标,将原子坐标转换成两两原子之间的欧几里德距离得到距离矩阵,这样就可以将三维坐标平面上的点转换为二维矩阵来表示。
服务器的输出
结构数据库的快速搜索算法的输出
在搜索算法模块,对于用户提交的单结构域结构和多结构域结构,服务器会自动选择在蛋白质结构域数据库或蛋白质链数据库中进行搜索。
link1
展示结果三个部分:
- 搜索得到的结果列表
- 查询结构和与该查询结构TM-score最高的10个结构的多重结构比对
- 描述查询结构每个残基位置对应非空格位置数量的 3D 图像
$$c_{i}=\frac{1}{N} \sum_{j=1}^{N} \delta_{i}(j)$$
$\delta_{i}(j)$是一个指示函数,查询结构的第 i 个残基与查找到的结果的第 j个残基匹配,则该函数值为 1,否则则为 0。每个残基的颜色从蓝到绿到红。若残基的保守性为 100%(即 C=1),则显示为蓝色;若残基的保守性为 0(即C=0),则显示为红色。
多重结构比对算法的输出
link2
展示结果三个部分:
- 结构之间的对应关系。
- 该比对的评价指标。
- 比对结果的3D比对结构图。
该部分通过 3Dmol 软件完成